
O teu país
Lorem ipsum dolor sit amet, consectetur adipiscing elit, sed do eiusmod
Lorem ipsum dolor sit amet, consectetur adipiscing elit, sed do eiusmod

Todos os trabalhos publicados foram gentilmente enviados por estudantes – se também quiseres contribuir para apoiar o nosso portal faz como o(a) Joana Vilela e envia também os teus trabalhos, resumos e apontamentos para o nosso mail: geral@notapositiva.com.
Trabalho em que é efetuada a análise da diversidade genética dos genes NEF2, LTA e GPR109B em populações humanas Europeias e Africanas...

As estatísticas sumárias da diversidade genética e os valores de Tajima D foram calculados no programa SLIDER. Os valores destas estatísticas para os 3 genes em cada população, podem ser consultados na tabela 1. Foram identificados os valores desviantes ao modelo de Wright-Fisher. O genes GPR109B (na população Europeia) e NFE2 (na população Africana) não evoluem de acordo com o modelo testado. Representações gráficas da distribuição do π (Pi) e do Tajima D podem ser observadas nas figuras 2 e 3. A distribuição do gráfico para o gene LTA não foi apresentada, já que nenhum dos valores para todas as populações estudadas são significativos. Para o gene NFE2, os valores de π (Pi) são positivos nos dois continentes, já os de Tajima D são negativos em África e oscilam entre valores positivos e negativos para a Europa. Em relação ao gene GPR109B os valores de π (Pi) são bastante positivos em ambos os continentes e os de Tajima D são maioritariamente positivos tanto na Europa como em África, chegando a atingir valores positivos bastante elevados.
Com base nos dados obtidos foram construídos gráficos do espectro de frequências folded para os 3 genes em ambos os continentes (Figura 1). Como se pode observar existe um excesso de alelos raros em África para o gene NFE2 e elevada quantidade de alelos de frequência intermédia para o gene GPR109B na Europa.
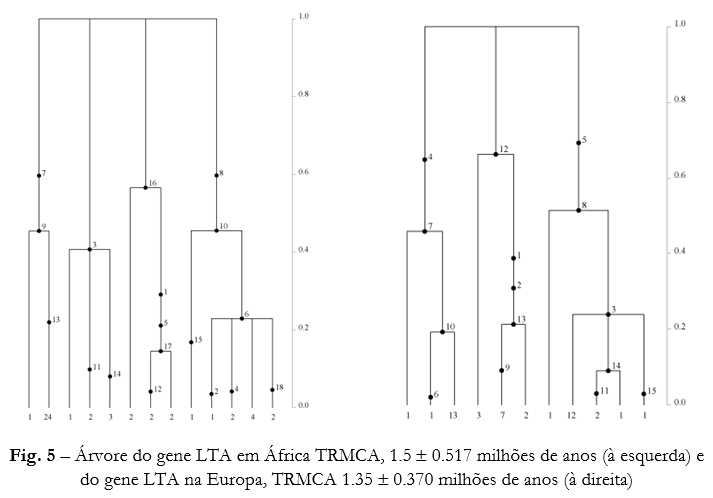
Através do programa GENETREE foram calculadas as estimativas do TMRCA para cada gene em cada população e reconstruiram-se as árvores para cada gene (Figuras 4, 5 e 6) . Para o gene NFE2 em África obteve-se uma árvore com uma estrutura em “pente” e com ramos terminais mais longos. Já a topologia da árvore obtida na Europa denota a existência de 2 linhagens diferenciadas que convergem mais recentemente. As árvores relativas ao gene LTA não exibem nenhum padrão específico. Para o gene GPR109B, ambas as árvores têm um aspecto semelhante, embora a diferenciação em 2 linhagens seja mais acentuada na árvore Europeia do que na Africana, que possui os ramos terminais ligeiramente mais longos e uma topologia mais próxima da estrutura em “pente”.
No programa MS, efectuaram-se simulações de diversos modelos demográficos, o que permitiu comparar os valores observados de Tajima D com as distribuições esperadas para os diferentes cenários testados. Para os genes das populações Africanas testaram-se modelos de expansão populacional e um Best fit-model com características ajustadas para estas populações. Para os genes da população Europeia foram testados 2 modelos de bottleneck com severidade e duração diferentes, 1 modelo de estruturação populacional e o Best fit-model mais adequado para esta população. Os valores significativos para todos os modelos testados podem ser consultados na tabela 1.




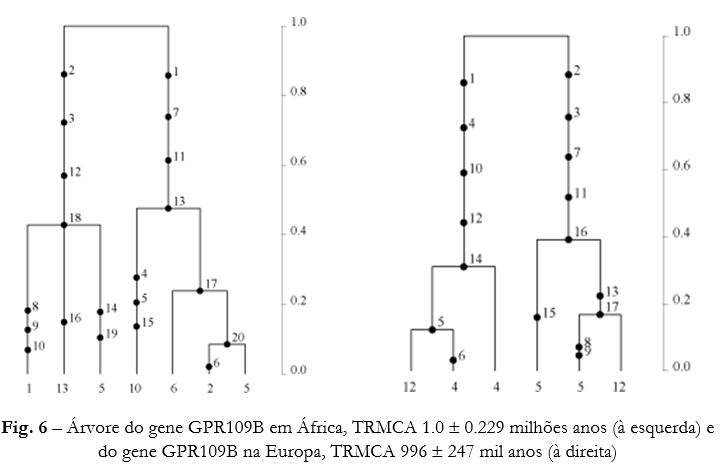

Como pode ser observado pela tabela 1 o gene GPR109B em África não tem o comportamento dentro dos valores esperados para o D de Tajima de acordo com o modelo de expansão populacional. Assim, este gene apresenta valores de Tajima D mais positivos do que o que seria de esperar para populações Africanas. O valor de Tajima D para a Europa apresenta-se bastante positivo em relação ao que seria de esperar. Dado que a demografia também não explica o padrão evolutivo deste gene para Europa, provavelmente estamos perante um cenário de selecção balanceada, já que o valor de π (Pi) é elevado revelando a manutenção de diversidade genética dentro da população. O número de posições variáveis (S) é também elevado. A topologia da árvore Europeia (Fig. 6) é concordante com um cenário de selecção balanceada, já que se distinguem 2 linhagens divergentes que persistem mantendo os níveis de diversidade na população. As coalescências são mais baixas do que em árvores típicas de expansão populacional ou de selecção positiva, e o número de mutações partilhadas é elevado. Este tipo de árvore representa uma população na qual existe uma elevada frequência de alelos intermédios.
Uma árvore com duas linhagens inicialmente diferenciadas que convergem mais recentemente é também encontrada para o gene NFE2 na Europa (Fig. 4). O valor de Tajima D pouco negativo exibido nesta população, não foi significativo para os modelos demográficos testados. Como tal, e de acordo com a topologia da árvore associada a este gene para o continente Europeu, podemos estar perante um cenário de subestruturação populacional ou de selecção balanceada. Em África, este gene apresenta um valor de Tajima D bastante negativo e significativo para o modelo de expansão populacional testado. Um cenário de selecção natural positiva poderá então explicar melhor os resultados para este gene, já que o valor de π (Pi) é reduzido. A topologia da árvore exibida por este gene em África (Fig. 4) apresenta uma configuração em forma de “pente”, com ramos terminais mais compridos, menor número de mutações partilhadas e indicadora de um elevado número de alelos de baixa frequência.
Através da análise dos gráficos do espectro de frequências (Fig. 1) é possível verificar que o gene NFE2 exibe um maior número de singletones tanto em África como na Europa, o que pode ser explicado pelo rápido aumento de alelos de baixa frequência num cenário de selecção positiva. O gene GPR109B apresenta um elevado número de alelos intermédios nos 2 continentes. Isto pode ser indicativo da presença de selecção balanceada já que nenhuma das situações pode ser explicada por processos demográficos, de acordo com os valores outliers de Tajima D. Os valores muito positivos de π (Pi) podem estar associados à manutenção de um certo nível de diversidade genética, já que este tipo de selecção não reduz drasticamente a variabilidade como é verificado num quadro de selecção positiva.
O cenário evolutivo para o gene GPR109B não pode ser explicado através de fenómenos demográficos, estando provavelmente sujeito à acção de selecção balanceada nos dois continentes. Relativamente ao gene NFE2 em África, os valores significativos de negativos de Tajima D indicam que poderá estar a ser afectado por selecção positiva. Na Europa este gene poderá estar a seguir uma evolução de acordo com um modelo de subestruturação populacional ou selecção balanceada. O gene LTA parece seguir um modelo de evolução genética neutral. O quadro de selecção balanceadora em África e na Europa para o gene GPR109B e de selecção positiva em África acompanhado de uma possível substruturação na Europa para o gene NFE2, não contradizem a teoria formulada pelo modelo “Out of Africa”.
 Angola
Angola Moçambique
Moçambique Cabo Verde
Cabo Verde Brasil
Brasil Inglês
Inglês Portugal
Portugal